



대신증권은 21일 한미약품에 대해 올 하반기 미국 식품의약국(FDA)의 현장 실사가 재개되면서 ‘롤론티스’와 ‘포지오티닙’의 신약 허가 가능성도 높아질 것으로 전망했다. 투자의견 ‘매수’와 목표주가 34만원을 유지했다.
임윤진 연구원은 “최근 FDA가 4월부터 해외 현장 실사를 본격적으로 재개할 것으로 발표한 가운데, 국내 코로나19 확진자 감소 시 하반기 실사 일정 확보가 가능할 것”이라며 “롤론티스 및 포지오티닙 두 약물 모두 생산시설 현장 실사가 최종 허가여부에 중요하게 작용할 것”으로 예상했다.
한미약품은 올해 안에 호중구감소증 치료제 롤론티스와 'HER2 엑손20' 변이 비소세포폐암 치료제 포지오티닙의 FDA 허가를 받는다는 목표다.
최근 한미약품의 협력사(파트너사)인 스펙트럼은 FDA에 롤론티스 신약허가신청서(BLA) 재제출을 완료했다. 지난해 8월 FDA로부터 보완요구서한(CRL)을 수령한지 7개월만이다.
이번 재제출은 앞선 CRL에서 시설 실사 요구가 포함됨에 따라 ‘2등급 재제출(Class 2 resubmission)’로 분류된다. FDA는 롤론티스 원료의약품(DS)을 생산하는 한미약품 평택 바이오공장 및 미국 완제의약품(DP) 위탁생산(CMO) 공장 현장을 실사할 예정이다.
임 연구원은 “30일 이내에 BLA 재제출 심사 등록(filing), 6개월 내 허가 여부 확인을 통해 이르면 올 9월 내 신약 허가가 가능할 것”으로 내다봤다.
이와 함께 스펙트럼은 지난달 11일 FDA에 치료 이력이 있는 HER2 엑손20 변이 비소세포폐암 환자 치료제로 포지오티닙의 신약승인신청서(NDA)를 제출했다. 해당 환자군에 대해 신속심사(패스트트랙) 지정을 받아, 허가신청자비용부담법(PDUFA) 일정인 오는 11월 24일에 최종 허가여부를 확인할 수 있을 것이란 예상이다.
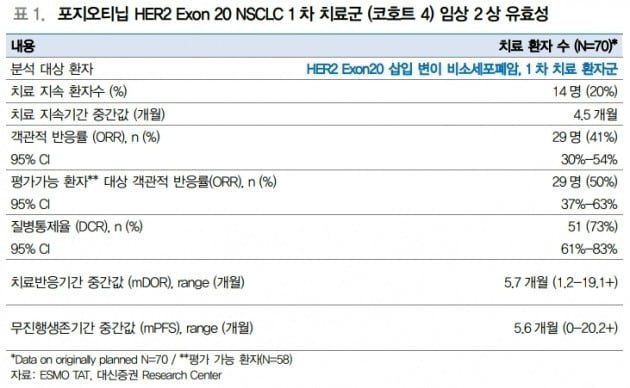
스펙트럼은 최근 표적항암요법 학술대회(ESMO TAT)에서 HER2 엑손20 변이 비소세포폐암 1차 치료 환자군 임상 2상(코호트 4)에 대한 긍정적인 중간결과를 발표했다. 전체 환자 70명의 객관적반응률(ORR)은 41%, 평가 가능한 환자군의 ORR은 50%로 나타나, 사전 정의된 ORR 하한값 20%를 넘어서며 1차 평가변수를 충족했다. 반응 지속기간(mDOR)은 5.7개월이었다. 이 중 환자 한 명은 치료 반응이 19개월까지 지속된 것으로 확인됐다.
임 연구원은 “현재까지 HER2 엑손20 변이 비소세포폐암 환자군에 대한 상용화된 치료제가 없는 가운데, 포지오티닙이 2차 치료제로서 허가될 가능성이 크다”며 “2차 치료제 대비 허가 기준이 높은 1차 치료제에 대해서는 3상 진행 후 허가를 신청할 것”이라고 했다.
이도희 기자
임윤진 연구원은 “최근 FDA가 4월부터 해외 현장 실사를 본격적으로 재개할 것으로 발표한 가운데, 국내 코로나19 확진자 감소 시 하반기 실사 일정 확보가 가능할 것”이라며 “롤론티스 및 포지오티닙 두 약물 모두 생산시설 현장 실사가 최종 허가여부에 중요하게 작용할 것”으로 예상했다.
한미약품은 올해 안에 호중구감소증 치료제 롤론티스와 'HER2 엑손20' 변이 비소세포폐암 치료제 포지오티닙의 FDA 허가를 받는다는 목표다.
최근 한미약품의 협력사(파트너사)인 스펙트럼은 FDA에 롤론티스 신약허가신청서(BLA) 재제출을 완료했다. 지난해 8월 FDA로부터 보완요구서한(CRL)을 수령한지 7개월만이다.
이번 재제출은 앞선 CRL에서 시설 실사 요구가 포함됨에 따라 ‘2등급 재제출(Class 2 resubmission)’로 분류된다. FDA는 롤론티스 원료의약품(DS)을 생산하는 한미약품 평택 바이오공장 및 미국 완제의약품(DP) 위탁생산(CMO) 공장 현장을 실사할 예정이다.
임 연구원은 “30일 이내에 BLA 재제출 심사 등록(filing), 6개월 내 허가 여부 확인을 통해 이르면 올 9월 내 신약 허가가 가능할 것”으로 내다봤다.
이와 함께 스펙트럼은 지난달 11일 FDA에 치료 이력이 있는 HER2 엑손20 변이 비소세포폐암 환자 치료제로 포지오티닙의 신약승인신청서(NDA)를 제출했다. 해당 환자군에 대해 신속심사(패스트트랙) 지정을 받아, 허가신청자비용부담법(PDUFA) 일정인 오는 11월 24일에 최종 허가여부를 확인할 수 있을 것이란 예상이다.
스펙트럼은 최근 표적항암요법 학술대회(ESMO TAT)에서 HER2 엑손20 변이 비소세포폐암 1차 치료 환자군 임상 2상(코호트 4)에 대한 긍정적인 중간결과를 발표했다. 전체 환자 70명의 객관적반응률(ORR)은 41%, 평가 가능한 환자군의 ORR은 50%로 나타나, 사전 정의된 ORR 하한값 20%를 넘어서며 1차 평가변수를 충족했다. 반응 지속기간(mDOR)은 5.7개월이었다. 이 중 환자 한 명은 치료 반응이 19개월까지 지속된 것으로 확인됐다.
임 연구원은 “현재까지 HER2 엑손20 변이 비소세포폐암 환자군에 대한 상용화된 치료제가 없는 가운데, 포지오티닙이 2차 치료제로서 허가될 가능성이 크다”며 “2차 치료제 대비 허가 기준이 높은 1차 치료제에 대해서는 3상 진행 후 허가를 신청할 것”이라고 했다.
이도희 기자
관련뉴스










